 Cофия Серда-Гонзалес (Sofia Cerda-Gonzalez], DVM, дипл. ACVIM (неврология)
Cофия Серда-Гонзалес (Sofia Cerda-Gonzalez], DVM, дипл. ACVIM (неврология)
Колледж ветеринарии Корнельского университета, Итака, США
Доктор Серда-Гонзалес окончила Корнельский университет в 2003 г., а затем прошла ротационную интернатуру в Нью-Йоркском Ветеринарном центре и резидентуру по неврологии и нейрохирургии в Университете Северной Каролины (200Д-2007 гг.). Свой диплом ACVIM по неврологии она получила в 2007 г. После этого она заняла должность доцента неврологии и нейрохирургии в Корнельском ветеринарном колледже, сконцентрировав свои научные исследования на аномалиях краниоцервикального сочленения.
КЛЮЧЕВЫЕ МОМЕНТЫ
Возраст животных, у которых диагностируют лизосомальные болезни накопления, составляет обычно меньше 1 года. Исключениями являются нейрональный цероидный липофусциноз и глобоидно-клеточная лейкодистрофия. При лизосомальных болезнях накопления простые методы осмотра и рутинные диагностические методы исследования (то есть анализы крови, рентгенография, УЗИ) могут предоставить значимую диагностическую информацию. Генетическое тестирование в некоторых случаях доступно для связанных с породой специфических заболеваний.
Введение
Дегенеративные наследственные заболевания, поражающие головной мозг, часто могут пугать из-за большого числа различных нарушений и того факта, что многие породы предрасположены к специфическим болезням. Однако если различные заболевания группируются в соответствии с их идентифицирующими характеристиками (то есть, по отличительным признакам), то разобраться с ними становится проще. В этой статье рассматриваются клинические характеристики лизосомальных болезней накопления, фокусируясь на моментах, помогающих идентифицировать эти заболевания у конкретного животного, а также обсуждаются методы, которые помогают в диагностике. В статье приведены сравнительные таблицы (таблицы 1 и 2) для описания различных дегенеративных энцефалопатий собак с клиническими признаками, совпадающими с лизосомальными болезнями накопления, особенно теми, которые наблюдаются у молодых животных и у предрасположенных к множественным дегенеративным расстройствам пород.
Отрицательный (то есть «нормальный») результат исследования на метаболические болезни не гарантирует отсутствия лизосомальных болезней накопления.
Этиология лизосомальных болезней накопления
Считается, что большая часть дегенеративных энцефалопатий, включая лизосомальные болезни накопления, возникает в результате врожденных метаболических перестроек, происходящих на клеточном уровне. Другие предполагаемые причины включают патологические изменения запрограммированной клеточной смерти и генетическую предрасположенность к аутоиммунному разрушению нейронов и/или глиальных клеток. Как подразумевает название, лизосомальные болезни накопления более специфичны и возникают в результате отсутствия или дисфункции лизосомальных ферментов. В нормальных условиях внутриклеточные материалы, например, компоненты клеточных мембран, разрушаются в лизосомах предсказуемым поэтапным путем с помощью последовательностей лизосомальных ферментов, позволяя конечным продуктам перерабатываться в новые клеточные материалы. Если какой-либо этап в этой цепи событий не может быть выполнен ввиду дефицита или отсутствия необходимого фермента, то образованный на предыдущем этапе материал ошибочно сохранится и начнет накапливаться, как правило, в форме сфероидов. Это накопление остаточного субстрата вызывает отек и токсическое действие на нейроны, поражая как нейроны, так и окружающие глиальные клетки. При большей части лизосомальных болезней накопления дисфункциональный этап включает кислую гидролазу, в особенности экзогликозидазы; в случае нейронального цероидного липофусциноза наблюдается недостаточность протеиназ (1-5).
Таблица 1. Проявления связанного с породой подострого наследственного некротизирующего энцефалита, который клинически напоминает лизосомальные болезни накопления
| Название заболевания | Типичный возраст дебюта | Клиническая картина | Прогрессирование | Диагностическиеисследования |
| L-2 гидроксиглутаровая ацидурия*(20) | 4-5 лет (4 месяца-7 лет) | Судороги, изменения поведения/сознания, мозжечковая атаксия, тремор головы | Скрытое начало, медленное прогрессирование | Повышение концентрации L-2-гидроксиглутаровой кислоты в плазме крови/моче |
| Мозжечковая кортикальная недостаточность* (21) | 4-6 лет (18 месяцев- 9 лет) | Мозжечково-вестибулярная | Медленное прогрессирование | МРТ, проба со стволовыми вызванными слуховыми потенциалами |
| Нейрональный цероидный липофусциноз (таламо- мозжечковая дегенерация с дебютом у взрослых) | 2-5 лет | Мозжечково-вестибулярная атаксия | Медленное прогрессирование | Исследование на цероидный липофусциноз собак (ЦЛС) |
| Подострая некротизирующая энцефалопатия стаффордширских бультерьеров (Лея- подобный синдром)* (23) | 7 месяцев- 2,5 года | Мозжечково-вестибулярная, косоглазие | Быстрое прогрессирование | Повышенная концентрация лактата, патологическое соотношение лактат: пируват |
| Подострая некротизирующая энцефалопатия аляскинского хаски (23,24) | 7 месяцев- 2,5 года | Мозжечково-вестибулярная атаксия, судороги, дефицит зрения, поведенческие изменения | Острое начало, затем статичное течение или улучшение; частые рецидивы | Отсутствуют |
| Подострая некротизирующая энцефалопатия йоркширских терьеров | 4 месяца-5 лет | Изменения сознания, зрительный дефицит, судороги, атаксия | Быстрое прогрессирование | Отсутствуют |
*Поражаемая порода — стаффордширский бультерьер.
Таблица 2. Дегенеративные поражения мозжечка у собак, клиническая картина которых аналогична лизосомальным болезням накопления
| Заболевание мозжечка | Породы, поражаемые в первую очередь | Другие поражаемые породы | Возраст дебюта | Сопутствующие симптомы |
| Мозжечковая кортикальная дегенерация/абиотрофия (1, 2, 21) | Керри-блю-терьер, стаффордширский терьер*, стаффордширский бультерьер; гордон-сеттер*, жесткошерстный бордер-колли, бретонский спаниель*, бульмастиф и староанглийская овчарка | Шотландский терьер, самоедская лайка, эрдельтерьер, лабрадор и золотистый ретривер, финский харьер, бигль, кокер-спаниель, дог, керн-терьер | 3-12 месяцев | Мозжечково-вестибулярные; прогрессирующие |
| Атаксия новорожденных (1,2) | Котон-де-тулеар | Неизвестно | Примерно 2 недели | Мозжечково-вестибулярные, «плавательные»; непрогрессирующие |
| Нейроаксональная дистрофия (1,2) | Ротвейлер | Колли, боксер, немецкая овчарка, чихуахуа | 1-2 года | Мозжечково-вестибулярные; прогрессирующие |
| Синдром печеночно-клеточной дегенерации (25) | Бернская овчарка | Неизвестно | 4-6 недель | Мозжечковая и печеночно-клеточная дегенерация; прогрессирующая |
| Гранулопривальная атаксия новорожденных (1,2) | Джек-рассел-терьер и парсон-рассел-терьер | Неизвестно | 1-2 месяца | Мозжечковая атаксия |
| Атаксия с поздним дебютом (1, 2) | Джек-рассел-терьер и парсон-рассел-терьер | Неизвестно | 6-9 месяцев | Мозжечковая дисфункция; прогрессирующие |
*Дебют у взрослых животных — 2-8 лет.
Таблица 3. Классификация лизосомальных болезней накопления
| Категория | Примеры болезней накопления |
| Гликопротеиноз | Фукозидоз; маннозидоз; нейрональный гликопротеиноз (болезнь Лафора) |
| Олигосахаридозы | Гликогеноз IA, II, IIIA и IVтипа |
| Сфинголипидозы | Ганглиозидоз GM1 I и II типа; глюкоцереброзидоз; глобоидно-клеточная лейкодистрофия |
| Мукополисахаридозы | Муколипидоз II типа; мукополисахаридоз I, II, IIIA и В, VI, VII типа |
| Протеиноз | Цероидный липофусциноз |
Лизосомальные болезни накопления, поражающие центральную нервную систему, можно сгруппировать по накапливаемым веществам (таблица 3). Для большей части лизосомальных болезней накопления предполагается аутосомнофецессивный механизм наследования, хотя для многих подтипов в качестве этиологии установлены специфические мутации (1-4).
Клиническая картина
Пораженные собаки рождаются, не имея симптомов, однако по мере накопления продуктов метаболизма у них постепенно развивается неврологическая симптоматика, которая склонна возникать постепенно и ухудшаться в течение недель или месяцев; иногда у собак развиваются острые ухудшения клинической картины, которые могут имитировать острый дебют заболевания, если предшествующие слабовыраженные изменения не были замечены (1).
Клинические признаки при большинстве лизосомальных болезней накопления появляются в течение первого или второго года жизни (то есть первые недели или месяцы). У собак с нейрональным цероидным липофусцинозом симптомы болезни обычно развиваются у молодых взрослых животных (то есть в возрасте 1-2 лет), хотя в редких случаях у животных не наблюдается симптоматики до зрелого возраста; описан поздний дебют заболевания в возрасте 9 лет. Аналогично время дебюта глобоидно-клеточной дистрофии в зависимости от породы может варьировать от 6 месяцев до 4 лет (1-3, 6-8), а в одной публикации описывается 14-летний шпиц с этим заболеванием (6).
Неврологически сначала отмечается нарушение функций мозжечка с вестибулярным компонентом или без такового. Мозжечковые признаки часто включают интенционный тремор, мозжечковую (то есть спастическую гиперметрическую) атаксию и позу с широко расставленными ногами. Также возможно выпадение рефлекса угрозы (без изменений зрения или дефицита лицевого нерва) и анизокория. После этого усиливается диффузное вовлечение центральной нервной системы, завершающееся переходом в глобальные/многоочаговые энцефалопатические симптомы. Ранние проявления отличаются при нейрональном цероидном липофусцинозе собак, фукозидозе и нейрональном гликопротеинозе (болезнь Лафора), которые сначала проявляются дисфункцией переднего мозга, судорогами, дефицитом зрения, а также изменениями поведения и сознания; в таких случаях мозжечковая дисфункция развивается позднее в ходе заболевания (1-4, 6-10).
Внечерепные проявления часто развиваются дополнительно к неврологической дисфункции. Они могут включать нарушение функций периферических нервов у собак при фукозидозе, глобоидноrлеточной лейкодистрофии, гликогензах и сфингомиелинозе. Пальпируемое увеличение локтевого нерва выявляется у собак с фукозидозом, что превосходно служит отличительным признаком заболевания (рисунок 1)( 1,2). Изменения скелета, например, карликовость, черепно-лицевые пороки, разболтанность и выпот в суставах развиваются у собак, страдающих ганглиозидозом, маннозидозом, мукополисахаридозом и муколипидозом II типа. При двух последних болезнях постоянный рост позвонков способен привести к сдавлению корешков нервов и внедрению костной ткани в спинной мозг, вызывающим компрессионную миелопатию и боль в позвоночнике. Напротив, ганглиозидоз проявляется патологическим расширением межпозвонковых щелей вместе с карликовостью и пороками развития лицевого скелета (11).
Лизосомальные болезни накопления могут иметь также более общие системные проявления. В частности, гепатоспленомегалия способна развиваться в случаях сфингомиелиноза, маннозидоза, гликогенза, глюкоцереброзидоза, ганглиозидоза и мукополисахаридоза. Кроме того, могут наблюдаться поражение сердца (при гликогенозе), дегенерация сетчатки (при цероидном липофусцинозе, муколипидозе III типа) и помутнение роговицы (при мукополисахаридозе I, VI, VII типа, ганглиозидозе, маннозидозе) (1,9).
Диагностика
Первоначальная клиническая гипотеза основывается на соответствии общей информации о животном и анамнезе, то есть молодом возрасте, породе и медленно прогрессирующей симптоматике, особенно при начальных проявлениях, указывающих на мозжечковую дисфункцию. Внечерепные проявления, например, увеличение локтевого нерва или черепно-лицевые пороки, укрепляют ветеринарного врача в первоначальных подозрениях.
Рисунок 1. Ветеринарный врач должен внимательно осмотреть конечности; увеличенный локтевой нерв может выявляться у собак с фукозидозом
Генетическое тестирование (то есть генотипирование) обеспечивает, при его доступности, наиболее быстрый и наименее инвазивный метод диагностики у животных с соответствующей клинической картиной. В большинстве случаев образцы (смыв со слизистой оболочки щеки) могут быть получены и подвергнуты исследованию непосредственно владельцами. К сожалению, эти тесты доступны только для подгруппы лизосомальных болезней накопления и для определенных пород, для которых идентифицированы вызывающие болезнь мутации. В частности, среди гликопротеинозов только для фукозидоза имеется генетический тест для английского спрингер-спаниеля. Среди сфинголипидозов в ряде стран выпускаются тесты для ганглиозидоза GM1 (португальская водяная собака), ганглиозидоза GM2 (японский хин) и глобоидно-клеточной лейкодистрофии (керн-терьер и вест-хайленд-уайт-терьер). Генетическая мутация, вызывающая гликогеноз IIIа типа (олигосахаридоз), может выявляться у курчавошерстного ретривера, тогда как генетическая мутация, вызывающая нейрональный цероидный липофусциноз, может обнаруживаться у бордер-колли, тибетского терьера, таксы, американского бульдога и английского сеттера. Наконец, генетическое тестирование возможно для мукополисахаридоза IIIB типа (шипперке), VI типа (миниатюрный пинчер) и VII типа (немецкая овчарка) (3, 9, 11-13).
В случаях, когда имеется подозрение на лизосомальную болезнь накопления, но либо отсутствует генетическое тестирование для пораженной породы, либо получен отрицательный результат теста, для окончательного диагноза требуется идентификация специфического продукта накопления или демонстрация недостаточности определенного фермента. Часто в таких случаях значимую информацию могут обеспечить рутинные методы исследования.
Физикальное исследование может выявить классические изменения, указывающие на заболевание, например, увеличение локтевого нерва, пороки развития лицевого черепа и разболтанность суставов. При пальпации живота и УЗИ органов брюшной полости можно выявить органомегалию, и если затем провести биопсию пораженных органов под УЗИ-контролем, то она может позволить выявить накапливаемый материал. В некоторых случаях простое исследование мазка крови выявляет патологически накапливаемый материал в лейкоцитах. Наконец, офтальмологическое исследование имеет значение для установления дегенерации сетчатки или изменений роговицы (1,5, 11-15).
Если при неврологическом исследовании обнаруживаются клинические признаки миелопатии, то рентгенография позвоночника служит эффективным методом исследования для выявления пролиферации костной ткани (которая наблюдается при мукополисахаридозе и муколипидозе II типа). В таких случаях наличие и распространенность сдавления спинного мозга могут подтверждаться с помощью магнитно-резонансной томографии (МРТ). Визуализирующие исследования (компьютерная томография и МРТ) также могут помочь в установлении предположительного диагноза, поскольку определенные болезни накопления способны иметь классические признаки. Например, собаки с нейрональsм цероидным липофусцинозом и мукополисахаридозом I типа демонстрируют цереброкортикальную атрофию и вторичную вентрикуломегалию; эти изменения наблюдаются также при когнитивной дисфункции у собак, однако если они выявляются у слишком молодых для когнитивной дисфункции животных, то это будет наводить на мысль о болезни накопления (рисунок 2) (1, 5,14). Дополнительно к цереброкортикальной атрофии у чихуахуа, страдающих нейрональным цероидным липофусцинозом, указывается присутствие утолщения мягкой мозговой оболочки (7). Напротив, у собак с глобоидно-клеточной лейкодистрофией и ганглиозидозом (GM1) МРТ может демонстрировать диффузное симметричное вовлечение белого вещества головного мозга; в таких случаях атрофия головного мозга развивается позднее (1,4, 10).
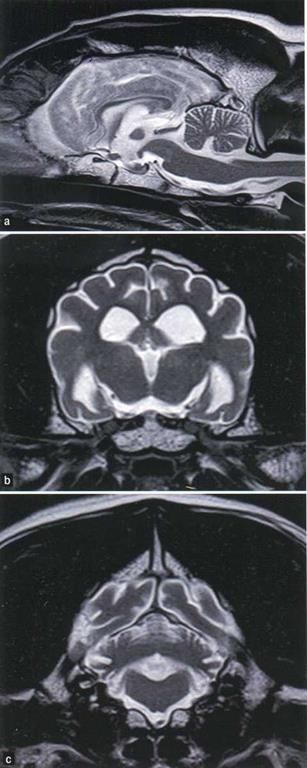
Рисунок 2. Т2-взвешенные МРТ-изображения головного мозга в саггитальной (а) и поперечной (Ь) ПЛОСКОСТИ, демонстрирующие атрофию коры головного мозга, межталамическое сращение и мозжечок у годовалой собаки смешанной породы с судорогами в анамнезе
Анализ ликвора, как правило, приносит нормальные результаты или демонстрирует неспецифическое небольшое повышение концентрации белка, однако он может быть эффективен в исключении других заболеваний, например, инфекционных или предполагаемого аутоиммунного энцефалита. Биопсия кожи и головного мозга может быть эффективной и надежной в идентификации продуктов накопления. Наконец, специфические «наборы анализов для метаболических заболеваний», в рамках которых анализируют образцы крови и мочи на наличие врожденных ошибок метаболизма, включая лизосомальные болезни накопления, позволяют установить окончательный диагноз. К сожалению, не все ошибки метаболизма могут быть идентифицированы. Например, среди лизосомальных болезней накопления только маннозидоз, мукополисахаридоз и ганглиозидоз могут быть выявлены в образцах мочи. По этой причине «обычный» метаболический набор анализов не исключает присутствия врожденных ошибок метаболизма (1, 2,12, 13).
Лечение
В целом эти состояния сопровождаются неблагоприятным прогнозом, поскольку клиническая картина прогрессирует, несмотря на попытки замедлить ее течение. Генная терапия (то есть доставка нормальной функциональной копии мутировавшего гена) находится на ранней стадии разработки, однако сопровождается многообещающими результатами, особенно в отношении а-маннозидоза кошек. Тот факт, что собаки могут имитировать множество лизосомальных болезней накопления человека, облегчает разработку лечения болезней накопления собак. Например, перенос гена через ликвор у биглей, страдающих мукополисахаридозом IIIA и VII типа, демонстрирует предварительные положительные результаты (16, 17). Интратекальное введение также позволяет эффективно доставлять рекомбинантный белок и приводит к снижению тяжести заболевания у собак, страдающих мукополисахаридозом 1 типа. Однако гемато-энцефалический барьер является основным препятствием к системному введению препаратов, и по этой причине требуются такие инвазивные методы, как интратекальное введение. Кроме того, в большинстве случаев методы контроля гуморального иммунного ответа на введение белка, доза и частота введения полностью определяются. По этой причине, насколько известно авторам, в настоящее время отсутствуют серийно выпускаемые средства лечения лизосомальных болезней накопления у собак(18, 19).
Литература
- ViteCH. Storage disorders. In: Vite CH, Braund KG. Braund’s neurology in small animals: localization, diagnosis, and treatment. 1st ed. Ithaca: International Veterinary Information Service, 2003 (www.ivis.org).
- Dewey CW. Encephalopathies: disorders of the brain. In: Dewey CW. A practical guide to canine&feline neurology. 2nd ed. Ames:Wiley-Blackwell, гоовщйго.
- Smith MO, Wenger DA, Hill SL, etal. Fucosidosis in a family of American-bred English springer spaniels. JAm Vet Med Assoc 1996;209:2088-2090.
- Hasegawa D, Yamato O, Nakamoto Y, etal. Serial MRI features of canine GM1 gangliosidosis: A possible imaging biomarker for diagnosis and progression of disease. Sci World J 2012:2012:1-10.
- Mizukami K, Kawamichi T, Koie H, etal. Neuronal ceroid lipofuscinosis in Border Collie dogs in Japan: clinical and molecular epidemiological study (2000-2011). Sci World J 2012:1 -7.
- Selcer E, Selcer R, Globoid cell leukodystrophy In two West Highland White terriers and one Pomeranian. Comp Cont Educ Pract Vet 1984;6:621 -624.
- Nakamoto Y, Yamato O, Uchida K, etal. Neuronal ceroid-lipofuscinosis in longhaired Chihuahuas: clinical, pathologic, and MRI findings. JAmAnim Hosp Assoc 2011 ;47:e64-70.
- Kondagari GS, Ramanathan P, Taylor R. Canine fucosidosis: A neuroprogressive disorder. Neurodegener Dis 2011 ;8:240-251.
- Mizukami K, Chang HS, Yabuki A, et al. Novel rapid genotyping assays for neuronal ceroid lipofuscinosis in Border Collie dogs and high frequency of the mutant allele in Japan. J VetDiag Invest 2011;23:1131-1139.
- Wenger DA, Victoria T, Rafi MA, etal. Globoid cell leukodystrophy in Cairn and West Highland White terriers. J Hered 1999;90:138-142.
- Yamato O, Masuoka Y, Yonemura M, etal. Clinical and clinico-pathologic characteristics of Shiba dogs with a deficiency of lysosomal acid beta- galactosidase: A canine model of human GM1 gangliosidosis. J Vet Med Sci 2003;65:213-217.
- University of Pennsylvania School of Veterinary Medicine Section of Medical Genetics Web site (PennGen). Available at: http://www.vet.upenn.edu/ penngen, Accessed Dec 28,2013.
- University of Prince Edward Island CIDD Database. Lysosomal storage diseases. Available at: ic.upei.ca/cidd/disorder/lysosomal-storage-diseases. Accessed Dec 28, 2013.
- ViteCH, Nestrasil I, Mlikotic A, etal. Features of brain MRI in dogs with treated and untreated mucopolysaccharidosis type I. Comp Med 2013;63:163-173.
- Keller CB, Lamarre J. Inherited lysosomal storage disease in an English springer spaniel. JAm Vet Med Assoc 1992;200:194-195.
- HaurigotV, Marco S, Ribera A. Whole body correction of mucopolysaccharidosis IIIA by intracerebrospinal fluid gene therapy. J Clin Invest 2013. Epub ahead of print.
- Xing EM, Knox VW, O’Donnell PA. The effect of neonatal gene therapy on skeletal manifestations in mucopolysaccharidosis VII dogs after a decade. Mol Genet Metab 2013;109:183-193.
- Hemsley KM, Hopwood JJ. Delivery of recombinant proteins via the cerebrospinal fluid as a therapy option for neurodegenerative lysosomal storage diseases. IntJ Clin Pharmacol Г/ier 2009;47 Suppl 1 :S118-123.
- Kondagari GS, King BM, Thompson PC, etal. Treatment of canine fucosidosis by intracisternal enzyme infusion. Exp Neurol 2011 ;230:218-226.
- Abramson CJ, PlattSR, Jakobs C, etal. L-2-hydroglutaricaciduira in Staffordshire Bull Terriers. J VetlntMed 2003;17:551-556.
- Kwiatkowska M, Pomianowski A, AdamiakZ, etal. Magnetic resonance imaging and brainstem auditory evoked responses in the diagnosis of cerebellar cortical degeneration in American Staffordshire terriers. Acta Vet Hung 2013;61:9-18.
- Abitbol M, Thibaud JL, Olby NJ, etal. A canine Arylsulfatase G (ARSG) mutation leading to a sulfatase deficiency is associated with neuronal ceroid lipofuscinosis. Proc Natl Acad Sci USA 2010;107:14775-14780.
- Collins D, Angles JM, Christodoulou J, etal. Severe subacute necrotizing encephalopathy (Leigh-like syndrome) in American Staffordshire Bull terrier dogs. J Comp Path 2013; 148:345-353.
- Brenner O, Wakshlag JJ. Alaskan Husky encephalopathy — a canine neurodegenerative disorder resembling subacute necrotizing encephalomyelopathy (Leigh syndrome). Acta Neuropathol 2000; 100:50-62.
- Carmichael KP, Miller M, Rawlings CA. Clinical, hematologic, and biochemical features of a syndrome in Bernese Mountain dogs characterized by hepatocerebellar degeneration. JAm Vet Med Assoc 1996;208:1277-1279.